Tech #856: Hybrid β-Hexosaminidase enzyme for gene and enzyme replacement therapy of GM2 Gangliosidoses
Tay-Sachs and Sandhoff diseases are severe lysosomal storage disorders caused by genetic defects in the subunits of the β-hexosaminidase (Hex) enzyme, leading to neurodegeneration due to GM2 ganglioside accumulation. To address challenges in enzyme replacement therapy, researchers at SickKids and the University of Manitoba have developed HexM, a novel hybrid Hex enzyme with improved stability and function, showing promise for treating these currently incurable diseases.
IP&C is seeking to license this technology to a biopharma partner to develop an enzyme replacement therapy or gene therapy for GM2 Gangliosidoses
Technology Reference Number
#856
Inventors
IP&C Contact
Publications
Patents
Granted: US 10400227 B2, EP 3119892 B1
Pending: CA 2940728 A1
Category
Therapeutics
Keywords
Hexosaminidase, GM2 Gangliosidoses, Tay Sach disease, Sandhoff disease, Gene therapy, Enzyme Replacement
Background
Lysosomal storage diseases are rare diseases caused by inborn errors of metabolism that result in enzyme deficiencies. GM2 gangliosidoses are one such group of lysosomal storage diseases, wherein a deficiency exists in the β-hexosaminidase (Hex) enzyme that breaks down fatty acid derivatives called glycosides in the lysosome to prevent their buildup in neuronal cells.
There are two major isozymes of Hex in human tissue: HexA (dimer of α and β subunits) and HexB (dimer of β subunits). Genetic defects in either gene encoding these subunits can result in the accumulation of GM2 ganglioside in neural tissue and either Tay- Sachs disease (defects in the α subunit) or Sandhoff disease (defects in the β subunit). Tay-Sachs and Sandhoff patients experience progressive neurodegeneration, mental and physical disabilities, including paralysis, seizures, vision and hearing loss, intellectual disability, and death in early childhood. The rate of disease progression and severity has been found to correlate with the level of residual Hex A activity: generally, clinical disease does not develop unless residual Hex A activity drops below a critical threshold of 5–10% of normal. There is no treatment available to cure Tay Sach or Sandhoff disease, and treatments are directed only to manage the symptoms.
Invention Description
The major impediment to establishing vector-mediated gene or enzyme replacement therapy (ERT) based on the β-hexosaminidase (Hex) enzyme is the need to synthesize both subunits.
To overcome this SickKids’s researchers in collaboration with researchers from the University of Manitoba have engineered a new artificial hybrid subunit of Hex (µ) combining α-subunit active site, the stable β-subunit interface and unique areas in each subunit needed to interact with GM2 activator protein – GM2 complex in vivo and form a new stable µ-dimer (HexM).
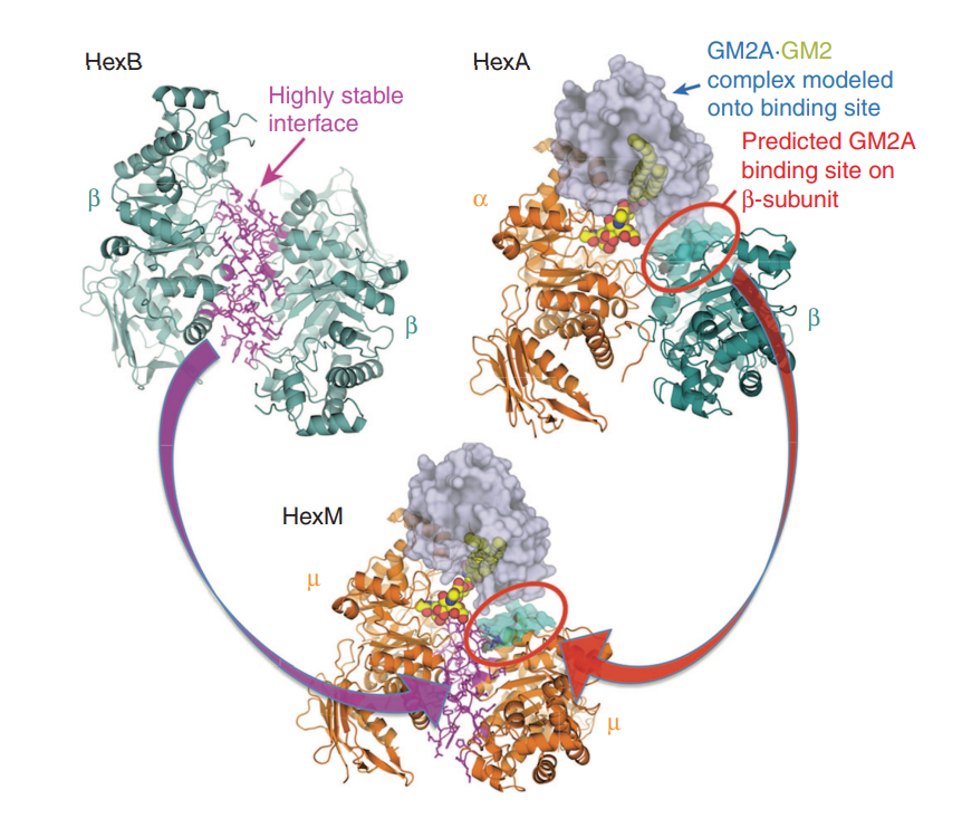
Figure 1.
Model of the active HexM quaternary complex. The μ-subunit of HexM is derived from the α-subunit (orange) of human HexA, modified to include the stable homodimer interface (magenta) formed between the β-subunits (teal) of human HexB and a region from the β-subunit predicted to interact with GM2AP. The GM2AP (grey) bound to GM2 (spheres) was modeled onto HexA.
The novel HexM hybrid enzyme offers promising potential for ERT in treating Tay-Sachs and Sandhoff diseases. It shows enhanced stability and efficient GM2 hydrolysis in vitro and in vivo.
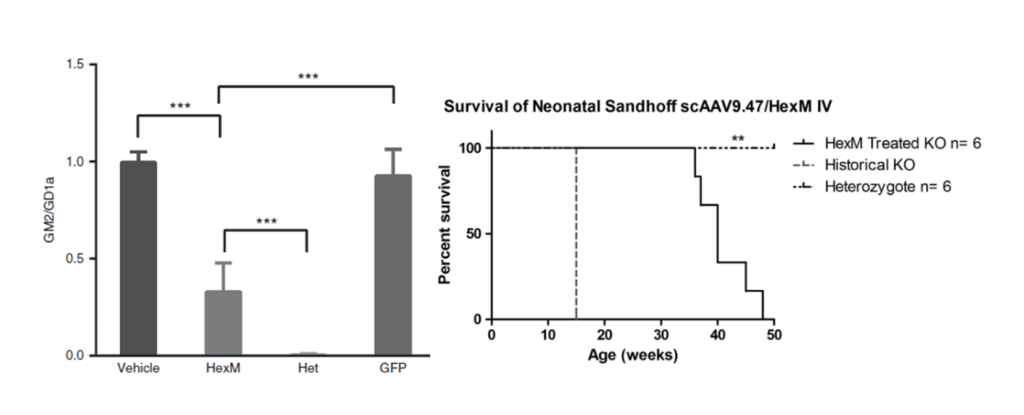
Figure 2.
GM2 levels are significantly reduced in the brains of Sandhoff disease mice 8 weeks after neonatal, intravenous injection of a AAV9.47-HEXM vector. Kaplan–Meyer survival curve depicting the HexM-treated group that outlived knockout (KO) age-matched controls. HexM-treated animals survived to a median age of 40 weeks, ranging between 36 and 49 weeks (n = 6). This extension in survival is significantly different compared with the historical 15-week humane endpoint (**p < 0.001).
Summary of Preclinical Data
In Vitro:
HexM secreted by engineered HEK cells was taken up by human cells lacking both HEXA and HEXB genes resulting in a 40-fold increase in GM2-hydrolyzing activity. This uptake was shown to be mannose-6-phosphate (M6P) dependent, confirming that HexM could utilize the natural lysosomal targeting pathway.
In Vivo:
Tay-Sachs Mouse Model: Intracranial injection of AAV vectors carrying HexM significantly reduced brain GM2 accumulation, matching or exceeding traditional HEXA-based therapies.Sandhoff Mouse Model: Neonatal intravenous HexM delivery lowered brain GM2 levels, despite modest increases in overall Hex activity.
Stability and Uptake:
HexM showed enhanced thermal stability and successful M6P-dependent uptake by Tay-Sachs patient fibroblasts, suggesting strong potential for enzyme replacement therapy.
Commercial Applications
The resulting size of the DNA-coding sequence for the new artificial subunit (µ) is well within the packaging capacity of adeno associated viral vectors to allow incorporation of the subunit and simplify vector mediated gene transfer approaches. This would enable in vitro transfection of a suitable cell for producing HexM and then isolating the enzyme for ERT or in vivo vector mediated gene therapy for treating GM2 Gangliosidoses.
The ability of HexM to be efficiently secreted and recaptured by deficient cells indicates strong potential for systemic enzyme replacement therapy targeting both peripheral tissues and, potentially, the CNS with advanced delivery techniques.
Licensing Opportunity
- Exclusive licensing opportunity in the field of enzyme replacement therapy
- Non-exclusive licensing opportunity in the field of gene therapy